Discussion Paper
Options for Possible Changes to the Patented Medicines Regulations, 1994 and the Excessive Price Guidelines
Released January 31, 2008
Table of Contents
- I Introduction & Purpose
- II Background
- III Overall Guidelines Review
- IV Options to Address Issues Arising from the Federal Court of Canada Decision
- Appendix A - Overview of the PMPRB Mandate
- Appendix B - PMPRB Patented Medicine Price Review Process
- Appendix C - Bibliography
I Introduction & Purpose
The Patented Medicine Prices Review Board (PMPRB) developed this discussion paper as part of the next phase of stakeholder consultations on the Board's approach in conducting reviews of the prices of patented medicines sold in Canada to ensure they are not excessive. This paper builds on the consultations launched in early 2006 regarding the relevance and appropriateness of the Board's Excessive Price Guidelines (Guidelines). It also builds on recent consultations with stakeholders regarding the decision of the Federal Court of Canada (FCC) in the matter of LEO Pharma Inc., regarding the calculation of the Average Price to be filed with the PMPRB.
This document is organized into two major sections, the first dealing with progress in regard to the issues identified in earlier consultations on the Board's overall Guidelines. The Board committed to further exploring several issues in its Stakeholder Communiqué of May 31, 2007, and is now seeking stakeholder input on certain proposed changes, and providing an update on work in progress on other issues.
The second major section deals with possible options to address the concerns of industry arising from the FCC decision, namely potential disincentives to the provision of “benefits” to customers arising from the non-discretionary methodology for calculating the Average Price of a patented medicine. This section includes options for changes to both the Patented Medicines Regulations, 1994 (Regulations) and the Guidelines. With respect to these particular options, the Board is seeking stakeholder feedback and has not yet taken a decision as to whether these options are appropriate to implement.
Your written feedback should be sent directly to Ms. Sylvie Dupont, Secretary of the Board, no later than March 3, 2008, at the following address:
Box L40
Standard Life Centre
333 Laurier Avenue West
Suite 1400
Ottawa, Ontario, K1P 1C1
Or by email: sdupont@pmprb-cepmb.gc.ca
If your comments are significant in length, please include a summary highlighting the key points of your submission. As with previous consultations, all submissions received by the Board will be posted on its Web site as part of the PMPRB's commitment to openness and transparency.
II Background
Overview of the Patented Medicine Prices Review Board
The Patented Medicine Prices Review Board (PMPRB) is a federal independent quasi-judicial body established in 1987 under the Patent Act (Act). The Minister of Health is responsible for the pharmaceutical provisions of the Act as set out in sections 79 to 103.
Although the PMPRB is part of the Health Portfolio1, it carries out its mandate at arm's-length from the Minister of Health. It also operates independently of other bodies such as Health Canada, which approves drugs for safety, quality and efficacy, and the federal/provincial/territorial public drug plans, which approve the listing of drugs on their respective formularies for the purposes of reimbursement for eligible beneficiaries. Please refer to Appendix A for a more detailed summary of the mandate of the PMPRB.
Review of the Excessive Price Guidelines2
Consultations on the Board's Guidelines began with the release of the Discussion Paper on Price Increases in 2005. The resulting feedback from stakeholders indicated that price increases in general were not their prime concern, but a number of other issues were raised concerning the appropriateness and relevance of the Guidelines as they pertain to the introductory price review process. The Board then sought views on a number of introductory price issues in a subsequent Discussion Guide released in May 2006, covering such matters as the categorization of new patented medicines, the approach to reviewing introductory prices, and whether to undertake price reviews in “any market.” This was followed by a series of face-to-face consultations in November 2006, which were held across Canada (Edmonton, Montréal, Toronto, Halifax and Ottawa). These consultations focussed on four issues, including the categorization of medicines and price reviews at the level of any market, and two new topics: whether and when to possibly “re-bench” (or “re-set”) a Maximum Non-Excessive (MNE) price, and guiding principles for the development of the Guidelines based on the pricing factors in the Act.
After careful consideration of all the comments it received, the Board issued a Stakeholder Communiqué on May 31, 2007, outlining its preliminary response on the issues put forward in the Discussion Guide and those raised during the consultation period, as well as on some additional matters, including: international therapeutic class comparisons, and the costs of making and marketing.
The Board then undertook a series of face-to-face bilateral consultations with stakeholder groups in September 2007, to hear sector-specific views on the proposed directions outlined in the Communiqué.
Work on many of the Guidelines issues is still under - way and will form the basis for further consultations in the spring of 2008. Within Section III of this Discussion Paper, the Board is seeking feedback from stakeholders on proposals pertaining to two issues discussed previously during the consultations:
1. Scenarios or triggers for when the Board should review prices for any relevant market and not just on a Canada-wide basis; and
2. Circumstances when it may be appropriate to deviate from the current CPI methodology and consider re-setting the MNE price for an existing medicine.
The Federal Court Decision in the Matter of LEO Pharma Inc.
In March 2007, in the midst of the more general review of the Guidelines, the FCC issued a decision in response to a judicial review application in the matter of LEO Pharma Inc. and the price of the patented medicine Dovobet.
In April 2007, the PMPRB issued a NEWSletter article informing stakeholders of the implications and impacts of the FCC decision.3 Stakeholders were instructed that all benefits (as defined by the Regulations in subsections 4(4) and 4(5); hereinafter referred to simply as “benefits”) must now be included in the calculation of the Average Price of a patented medicine. It was also explained that the FCC decision superseded the direction provided by the Board in its April 2000 NEWSletter and Chapter 1, subsections 5.3 and 5.4 of the Board's Guidelines, that had permitted discretion on the part of patentees to include or exclude certain benefits (related to compassionate use programs, trial prescription programs and expenditure limitation agreements) as long as the inclusion or exclusion thereof was consistent in all reporting periods.
Significant concern was expressed by the patented pharmaceutical industry, regarding the potential disincentives the decision would have on the willingness of companies to offer, or continue to provide, various benefits to their customers.
Representatives of the innovative pharmaceutical and biotechnology industries were given the opportunity to comment on the implications of the FCC decision during specific bilateral meetings with the Board during the summer of 2007. These sectors also offered further views on the FCC decision as part of the Board's bilateral meetings with stakeholder groups on the overall Guidelines review held in September 2007. Some also provided further comments in submissions pursuant to the second pre-publication of proposed regulatory changes published in Canada Gazette, Part I, on October 6, 2007.4
To gain further insight into specific industry issues, Board Staff met with representatives from Canada's Research-Based Pharmaceutical Companies (Rx&D) and BIOTECanada throughout the fall of 2007.
The PMPRB issued a Stakeholder Communiqué on October 18, 2007, stating that the work to address these concerns will take some time to complete, so the Board will not require any change in the manner in which the Average Price is calculated for the three periods (July-December 2007; January-June 2008; July-December 2008); that is, patentees may elect to include or exclude all benefits and reductions in the calculation of Average Prices, as long as consistency with previous reporting periods is maintained.
The Board is legally obligated to abide by the Regulations and the decisions of the FCC, and is therefore considering potential changes to both the Regulations and the Guidelines that would be consistent with its statutory responsibility to ensure that the prices of patented medicines sold in Canada are not excessive, but at the same time would not unduly create disincentives relative to benefits for customers. Therefore, Section IV of the document outlines a range of possible options relative to the Regulations and the Guidelines which could possibly mitigate the issues arising from the FCC decision and/or other future difficulties by clarifying or excluding certain benefits in the Average Price calculation.
It should be noted that any changes to the Regulations would be applicable to all patentees and all medicines under the Board's jurisdiction. Please note that while the Board is seeking stakeholder views on these options, it has yet to determine which, if any, it will implement. Finally, please note that the options put forward in this discussion paper in some cases are not mutually exclusive and could potentially be implemented simultaneously.
III Overall Guidelines Review
A. Proposed Scenarios for Consultation
i) Any Market Price Review
Background
In the event that the Board finds that a price of a patented medicine sold in Canada is excessive, it can order a price reduction. Section 83 of the Act provides that the Board may make such a finding and order in respect of the price at which a patented medicine is being sold in any market in Canada (emphasis added).
Currently, the PMPRB typically uses the Average Price for Canada as a whole to conduct the various price tests during price review. The Average Price is calculated for each drug product (identified at the level of the Drug Identification Number or DIN5) by dividing the total net revenue (i.e., the sum of revenues from each class of customer in each province/territory) by the total units sold (i.e., to all classes of customer in each province/territory).
The Discussion Guide released in May 2006 showed that while the Average Price for some drugs in Canada are considered to be within the Guidelines, the Average Price within some markets (i.e., class of customer or province/territory) did vary over 25% above the MNE price. As a result, some stakeholders are concerned that if some provinces/ territories and/or classes of customer negotiate price concessions below the MNE price, the offset may be that other provinces/territories and/or classes of customer may pay higher prices (above the MNE price).
Views of Stakeholders
In the 2006 and 2007 consultations, stakeholders expressed the view that, if price reviews are conducted at the level of any market, they should be undertaken, on a case-by-case basis, where appropriate.
Board Response
In its May 31, 2007, Stakeholder Communiqué the Board agreed with this approach and committed to identifying circumstances where it may be appropriate to review prices in any market in Canada.
Proposal
The Board seeks comments on the following proposed circumstances when a price review at the level of any market would be conducted.
- At introduction (during the period of first sale of a medicine in Canada), the PMPRB will ensure that the Average Price for all markets (i.e., for each class of customer and for each province/ territory) does not exceed the MNE price.
- In future years, if the Average Price for Canada appears to exceed the MNE price in any period, as part of the investigation Board Staff will review the price for each class of customer and each province/territory to determine in which market(s) the price appears to be excessive.
- If a patentee enters into a Voluntary Compliance Undertaking (VCU), or is subject to a Board order following a public hearing, the PMPRB will review prices in each market (i.e., each class of customer and each province/territory) for all reporting periods covered by the VCU or order to ensure that the price in any market does not exceed the MNE price.
- Any substantiated complaint of apparent excessive prices in any market will be investigated.
These scenarios are designed to be transparent as to when Board Staff would undertake an any market price review on a case-by-case basis. However, this should not be construed as limiting the Board's authority, pursuant to the Act, to review the price of a medicine in any market in Canada under any circumstance where it deems such a review to be relevant.
ii) Re-Setting the MNE Price
Background
The price of a patented medicine is reviewed when it is first sold in Canada to determine the introductory non-excessive benchmark price. The review is based on the indication of the medicine, the clinical evidence available at that time regarding therapeutic improvement and the appropriate price test (depending on the category of the medicine). Thereafter, the MNE price is determined as the lower of the price increase allowed by the CPI-adjustment methodology and the highest international price.
The Guidelines currently provide for two cases where the MNE price for existing drugs may be revisited and a new MNE price may be set (referred to as “re-setting”, formerly “re-benching”, the MNE price):
1. Investigational New Drugs and Special Access Programme
When a drug product is sold as an Investigational New Drug (IND) or under the Special Access Programme (SAP), the Guidelines state that it may be appropriate to adjust the price when the drug is granted a Notice of Compliance (NOC) although guidance on when this should occur is not given; and>
2. Patented drug products sold in less than 5 comparator countries6
When the pivotal introductory price test for a drug product is the Median of the International Price Comparison Test and the drug is sold in less than 5 countries during the introductory period, the Guidelines say that the price may be reviewed at the end of 3 years or when the medicine is sold in at least 5 countries, whichever comes first.
Views of Stakeholders
During the stakeholder consultations throughout 2006 and 2007, the Board asked participants if, and under what circumstances, the PMPRB should consider re-setting the MNE price.
In general, stakeholders expressed a range of positions regarding whether and when to re-set the MNE price. Representatives of the patented pharmaceutical industry did not support the idea, since the prospect of re-setting the MNE price would create commercial uncertainty. On the other hand, other industry stakeholders felt that there could be circumstances when price re-setting would be appropriate to consider, but this should be done on a case-by-case basis. Non-industry stakeholders expressed the importance of taking new scientific evidence into consideration as it becomes available and suggested that, in the future, such circumstances should be aligned with Health Canada's proposed Progressive Licensing initiative. There was consensus among stakeholders that, whatever criteria and processes are established for re-setting the MNE price, they should be clear, transparent and not overly burdensome on either the PMPRB or the patentee.
Board Response
After consideration of all comments and feedback, the Board expressed in its May 31, 2007,Stakeholder Communiqué, that it would be appropriate to give further consideration to additional circumstances where re-setting the MNE price may be undertaken.
Proposal
The Board seeks comments on the following proposed circumstances when it would be appropriate to consider re-setting the MNE price on a case-bycase basis. Further work would be required on implementation issues such as the transitional period to comply with the re-set MNE price.
1. When the MNE price can be shown to not cover the patentee's cost of making and marketing the drug.
The following are descriptions of three scenarios under a cost rationale that could arise and are proposed to be included in the Guidelines:
i) When a drug product that has been sold as an Investigational New Drug (IND) or under the Special Access Programme (SAP) at an artificially low price is actually approved for sale in Canada and launched on the Canadian market, and it can be shown that the actual costs of making and marketing the approved drug product are higher than the MNE price allowed by the current existing price tests;
ii) When a new government regulation or policy requirement imposes additional costs on the patentee and the MNE price of the drug at that time would not cover the increased total costs of making and marketing the medicine; and
iii) When an ongoing shortage (the length of shortage that would warrant consideration is still to be determined) of the drug ingredient increases the acquisition cost of the ingredient leading to increased costs of making and marketing the medicine above the MNE price.
For each scenario, supporting evidence would be required, but has yet to be determined in part due to ongoing work to determine what activities and costs would be eligible under a definition of “making” and “marketing.”
Assuming the patentee is successful in providing the evidence needed to support a re-setting of the MNE price based on costs, the price re-setting could be done by:
a) Re-performing the original price test in the current year to arrive at a new MNE price for the medicine; or
b) Accepting the current Average Price of the medicine as being non-excessive, thereby re-setting the MNE price for that year.
2. When the scientific information/evidence available at the time the medicine was first introduced was not sufficient to determine with confidence its category of therapeutic improvement, or when new post-market evidence suggests the initial categorization was inappropriate.
The following are descriptions of three scenarios when the scientific information/evidence in the introductory period might not be sufficient:
i) A drug product is being sold as an Investigational New Drug (IND) or under the Special Access Programme (SAP) and proper and sufficiently robust clinical trials have not been completed or are unavailable;
ii) A Notice of Compliance with conditions (NOC/c)7 has been granted but Health Canada has specified further research to be undertaken post-market, to confirm health outcome improvements;
iii) A drug is indicated for a rare, life-threatening disease and the scientific evidence is very limited because the patient population is too small to conduct proper and sufficiently robust clinical trials.
It could be that, after being sold in Canada for say 3 to 5 years, additional clinical trials and/or post market surveillance may provide new evidence to better determine the relative category of therapeutic improvement of the medicine. Re-setting the MNE price would recognize the real value of the medicine. Rather than develop its own review cycle, it has been proposed that the PMPRB adopt a regulatory life-cycle approach in line with the Progressive Licensing initiative of Health Canada.
3. When the Median of the International Price Comparison is the pivotal test and the medicine is sold in too few countries at introduction.
The current Guidelines state that an “interim” price will be used in cases when a medicine is sold in fewer than 5 countries at the time of its introduction. The interim price may then be reviewed at the end of 3 years or when the medicine is sold in at least 5 countries, whichever comes first.
In reviewing the Guidelines, the threshold of 3 years, or less than 5 countries appears somewhat arbitrary. It would be plausible to determine a fairly representative median international price based on the prices of the medicine in as few as 3 countries. Similarly, it does not appear that the 3 year timeframe limit is based on evidence of the general lag time in the introduction of medicines in various world markets.
The Board is seeking feedback on a possible change to the number of countries necessary to initiate a re-review, and the timeframe for the re-review.
Number of Countries
- It is proposed that the re-review of the Median of the International Price Comparison Test be undertaken when the medicine is sold in at least 3 countries (if it was originally sold in fewer than 3 countries).
Timeframe for Re-Review
The PMPRB is seeking stakeholder comment on three possible options regarding the timeframe for revisiting the Median of the International Price Comparison Test for a medicine with an interim price:
i) Maintain the status quo for the timeframe, where the Guidelines would retain the criteria that the interim price be reviewed at the end of 3 years or when the medicine is sold in at least 3 countries (assuming the Board's proposal that the minimum number of countries be decreased to 3), whichever comes first; or
ii) Maintain the existing timeframe of 3 years, but in the future, align it with the timeframes adopted under Health Canada's proposed Progressive Licensing initiative, when implemented; or
iii) Eliminate a time limit altogether and re-review the interim price of the medicine when it is sold in at least 3 countries, no matter how many years from date of first sale this may be.
B. Updates on Other Issues Under Guidelines Review
The PMPRB is continuing its work on a number of other issues raised during the course of the Guidelines review, as described below.
i) Principles
Background
The authorities granted in the Act and its associated Regulations are put into operation by way of the Guidelines. However, it is not always clear what principles may have guided the Board in extrapolating the requirements of the Act into the Guidelines.
Stakeholder Views
During the consultations, stakeholders linked a wide variety of principles to the Board's mandate, such as lowest reasonable price, price stability, price predictability, to name a few.
Board Response
The Board is cognizant that the Government's objective in creating the PMPRB was to ensure the additional patent protection provided to pharmaceutical patentees stemming from changes in the Act did not translate into excessive prices. In keeping with this objective, the Board's mandate is to ensure that prices charged by patentees for patented medicines sold in Canada are not excessive, thus protecting the interests of consumers. The Board intends to include language to this effect in the preamble to the Guidelines.
Status
Once the Board completes its analysis and consultations on directional changes to the Guidelines, revisions to the actual Guidelines text will be drafted. At that time, the preamble will be updated to reflect the above. The Board intends to issue proposed language for revisions to the preamble of the Guidelines for stakeholder notice and comment in the spring or summer of 2008.
ii) Categories of Medicines
Background
The price review process for all new drugs begins with a scientific review. The current Guidelines establish three categories for new patented drug products for the purpose of introductory price reviews (i.e., line extension; breakthrough or substantial improvement; and moderate, little or no improvement).
Stakeholder Views
Some stakeholders believe the current system of categorizing drugs does not recognize incremental innovation. Some stakeholders suggested separating “moderate improvement” from “little/no improvement.” Others suggested additional factors be considered, such as improvement in patient compliance and ease of use. Still others suggested that the categories be eliminated altogether.
Board Response
The Board believes that some assessment of therapeutic value is needed and work on options for possible revisions to the current approach is appropriate. To this end, the Board will establish a Working Group whose mandate will be to examine the possibility of developing definitions or parameters relating to “breakthrough/substantial improvement”, “moderate improvement” and “little or no improvement,” along with supporting evidence requirements.
Status
The Board's Working Group on Therapeutic Improvement was established in October, 2007. The terms of reference for the Working Group can be found on the Board's Web site. The Working Group is expected to deliver its final recommendations to the Board in March 2008.
iii) International Therapeutic Class Comparison
Background
The Act says that the Board shall take into consideration the prices at which other medicines in the same therapeutic class have been sold in countries other than Canada, but there is no reference whatsoever to this mandatory price factor in the Guidelines.
Stakeholder Views
Stakeholders expressed support for exploring the use of international therapeutic class comparators in the price review process, but held mixed positions on how and when such international prices and comparators should be taken into consideration.
Board Response
The Board recognizes that this is not a factor that is described in its Guidelines. As a first step, the Board will establish a small group of experts to develop a methodology for identifying appropriate therapeutically comparable medicines in comparator countries. The focus of the mandate for this group of experts will be based on scientific and clinical considerations only and will not include work on possible price tests nor when or how this factor may be incorporated in price tests.
Status
The Working Group on International Therapeutic Class Comparison was established in November 2007. The terms of reference for the Working Group can be found on the Board's Web site. The final report and recommendations of the Working Group are expected to be delivered to the Board in March 2008.
iv) Price Tests
Background
The Board began its comprehensive review of the Guidelines in follow-up to comments by stakeholders that introductory prices of patented medicines were high.
Stakeholder Views
The views of various stakeholders are wide ranging, and depending upon the stakeholder's perspective, this can mean that the current tests undervalue or overvalue patented innovations.
Board Response
In May 2007, the Board reserved comment on price tests in general and their use, in light of the decision to establish Working Groups on Therapeutic Improvement and the International Therapeutic Class Comparison.
Status
The Board will establish a Price Test Working Group to review the appropriateness of the current price tests once the Board has reviewed the advice of the two Working Groups and taken a decision on how it proposes to proceed on the matter of domestic categories of medicines and international comparators. It is anticipated that the first meeting of the Working Group will take place in April 2008.
v) Costs of Making and Marketing a Medicine
Background
Pursuant to subsection 85(2) of the Act, where after taking into consideration the factors referred to in subsection 85(1), the Board is unable to determine whether the medicine is being or has been sold in any market in Canada at an excessive price, the Board may consider the costs of making and marketing a patented medicine in determining whether or not its price is excessive.
Stakeholder Views
Industry saw little merit in pursuing this matter as it would probably never be used. Others thought there was value in defining making and marketing costs, but recognized that it could be difficult.
Board Response
While, to date, the Board has not had to give consideration to subsection 85(2) to make a determination of excessive pricing, it recognizes this situation could arise. As a result, the Board will be considering specific circumstances where it may be appropriate to consider these costs. It will also be seeking input from experts and stakeholders on how making and marketing should be defined, what type of cost evidence would be needed, as well as what would be considered appropriate sources of such evidence.
Status
Consultants are being engaged by the Board to carry out analyses from two perspectives — an economics perspective and an accounting perspective. A Working Group has been formed and will provide comments on the draft papers of the consultants and any other overall views and advice. The consultants' final reports are due in April 2008.
vi) Price Increases (CPI Methodology)
Background
The current CPI methodology was developed as a result of extensive consultation with all stakeholders in 1992 and 1993. In 2005, as a result of reports by third parties regarding price increases, the Board released a Discussion Paper to further hear stakeholders' views on such increases. For the most part, respondents said that price increases were not the main issue; rather, of more concern were introductory drug prices. In the interest of the completeness of its review of the Guidelines, the Board has also undertaken an assessment of its CPI methodology.
Stakeholder Views
During the consultations, stakeholders said that the CPI methodology should be more flexible.
Board Response
As a result of extensive consultations that took place in 1992-93, the current CPI Guidelines were developed: price increases were limited to the cumulative change in the CPI over three years, and any price increase in a given year could not exceed 1.5 times the forecast change in the actual CPI. However, the methodology can, for example, result in rare circumstances where the MNE price calculated for the year under review is less than or only equal to the Average Price of the previous year which was within the Guidelines. The Board does not believe this was the intent of the methodology. The Board will be drafting language to permit some flexibility in applying the existing CPI methodology for comment by stakeholders.
Status
The Board has temporarily suspended work on changes to the CPI-adjustment methodology to address these rare circumstances where no price increase might be allowed, pending feedback from stakeholders on new options in the next section of the Discussion Paper pertaining to further options for changes to the Guidelines that pertain to the CPI-Methodology used to determine the MNE price.
IV Options to Address Issues Arising from the Federal Court of Canada Decision
This section outlines a range of possible options for changes to the Regulations and/or the Guidelines, designed to mitigate concerns arising from the Federal Court of Canada (FCC) decision. It should be noted that any changes to the Regulations and/or the Guidelines would be applicable to all patentees and all medicines under the Board's jurisdiction. Please also note that the Board has yet to determine which of the following options, if any, it will implement.
A. Regulatory Options
Option 1 Maintain the current Regulations and respect the outcome of the FCC decision.
Description
The existing Regulations would apply, taking into account the FCC decision. This would mean that patentees would be required to include all benefits listed in the Regulations in the calculation of a medicine's Average Price, whether or not they are provided under a compassionate release program, trial prescription program, expenditure limitation agreement or pursuant to any other initiative.
Rationale
It could be argued that the original intent of the Regulations was that a true Average Price be calculated and therefore selectively removing certain benefits from the reported Average Price is inappropriate. Furthermore, it could be argued that, pursuant to the Act, the Board does have the necessary flexibility to determine the MNE price in such a way so as not to penalize patentees that offer benefits.
Analysis
This option, considered in isolation of the other options, would not resolve the possible disincentives to provide benefits to customers. If, however, this option were to be combined with the administrative options put forward under the further Guidelines options in the next section, patentees' concerns about the MNE price being severely constrained by a decreased Average Price (due to the inclusion of all benefits) would likely be mitigated. This option would also not create any significant bias in the reporting of actual pharmaceutical trends by the PMPRB, in that all benefits would be fully accounted for in the Average Price.
Option 2 Amend the Regulations to exempt patentees from the requirement to report benefits (payments) provided to third-party payers (F/P/T drug plans and potentially private insurers if similar payments are negotiated in the future).
Description
This regulatory option would seek to modify the list of benefits contained in subsections 4(4) and 4(5) that must be included in the calculation of the Average Price, by specifically stating that payments to third-party payers are not “benefits” that must be reported.
Rationale
The PMPRB's mandate pertains to regulating the prices that patentees charge – the factory-gate price – for prescription and non-prescription patented drugs sold in Canada to all classes of customer, in each province and territory, for human and veterinary use. While not defined in the Regulations, the Guidelines specify four classes of customer (pharmacy, hospital, wholesaler and other). It could be argued that, third-party payers are one step removed from typical customers. It could also be argued that, if the price at which the medicine is sold to the pharmacy is not excessive, then the PMPRB should not be concerned if a payment made later to a province effectively further reduces the price.
Analysis
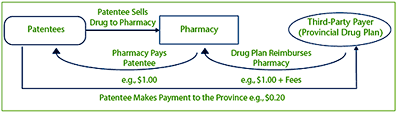
Expenditure limitation agreements work to limit the total cost to the payer. For example, the patentee sells a medicine to the pharmacy. The pharmacy is reimbursed the cost of the medicine (including retail mark-ups and fees) by the drug plan. As part of the drug benefit plan's agreement to list the medicine on its formulary, the patentee enters into an expenditure limitation agreement and agrees to provide a payment directly to the province/territory to compensate in part for the cost of the medicine that was reimbursed by the third-party payer.
The PMPRB's mandate is to ensure that the “factorygate price” is not excessive. While the initial price at which the medicine in this example is sold to the pharmacy is $1.00, the issue is whether this is the Average Price that should be regulated, or should it be $0.80 (taking into account the $0.20 payment made by the patentee to the drug plan)?
Such payment arrangements may not be so simple and may, for example, be negotiated on the basis of other factors such as the achievement of target improvements in health outcomes. The agreement may also involve multiple drugs, both patented and non-patented.
At the moment, such expenditure limitation agreements appear to exist or be under discussion relative to only a few drug benefit plans. The Transparent Drug System for Patients Act, 2006, in Ontario enables the Executive Officer to negotiate agreements with pharmaceutical companies related to drug benefit list prices and payments to the province under the public drug program.
The Régie de l'assurance maladie du Québec (RAMQ – Québec's public drug benefit program) has a policy that requires drug companies to offer it the best price provided to any other provincial drug benefit program in Canada. In addition, the new legislation (Bill 130) gives the Minister the authority to enter into agreements with drug manufacturers either as financial risk sharing for specific medications or compensatory measures to mitigate the negative impact of a price increase on a drug plan. It is likely that other public and potentially even private, drug plans will, in the future implement similar legislation and/or policies.
Provincial and territorial drug benefit programs make up a large portion of drug expenditures. In 2006, Ontario and Québec's drug benefit programs accounted for 40.8% and 36.1% respectively of total provincial drug expenditures. At the national level, in 2006 all provincial and territorial public drug benefit program expenditures accounted for 38.4% of all drug expenditures in Canada.
Other jurisdictions have expressed concern that if one jurisdiction enters into an arrangement, which reduces the price paid by the drug plan, other jurisdictions may pay higher prices to offset the lost revenue for the patentee. Some stakeholders may view this as a circumstance in which the review should include verification that the price is not excessive in any market.
It is also worth noting that the PMPRB has a reporting mandate to publish trends in pharmaceutical pricing. It could be argued that such reporting would become less representative of the pharmaceutical market if these payments to third-party payers are excluded. Others would argue that off-invoice rebates are generally not captured in price information gathered by other organizations, for example, IMS Health.
Figure 1
Depiction of a Sample Expenditure Limitation Agreement and the Flow of Payments between Parties
Option 3 Amend the Regulations with respect to free goods.
There are three variations on which the Board is seeking comments.
i. Amend the Regulations to exclude all free goods from the calculation of the Average Price.
Description
This regulatory option would remove the reference to “free goods” in subsections 4(4) and 4(5), thereby excluding all free goods from the calculation of the Average Price.
Rationale
The statutory mandate of the Board pertains to patented medicines sold in Canada. If a quantity of medicine is provided free of charge and without consideration, it can be argued that it should not be within the Board's jurisdiction, as these quantities do not fall under the definition of a “sale.”
Analysis
The exclusion of all free goods from the calculation of the Average Price would ultimately result in a higher Average Price for those patentees currently reporting free goods. For those including a substantial amount of free goods in the calculation of the Average Price, their exclusion might even cause the Average Price to become excessive under the existing Guidelines.
ii. Amend the Regulations to exclude free goods from the calculation of the Average Price when only free goods are provided to a particular customer class.s
Description
This regulatory option would amend the reference to “free goods” in subsections 4(4) and 4(5) to exclude from the calculation of the Average Price free goods provided to a particular customer class, if all goods received by that class are free.
Rationale
The statutory mandate of the Board pertains to patented medicines sold in Canada. If a patented drug is not actually sold by a patentee to a particular customer class, but is provided free of charge and without consideration, it can be argued that it should not be within the Board's jurisdiction. For classes that receive some medicine free of charge but pay for other quantities, the free portion may be considered more a form of discount for the customer (e.g., buy one, get one free).
Analysis
The exclusion of goods that are provided exclusively on a free basis to a particular customer class from the calculation of the Average Price, would ultimately result in a higher Average Price for that class and when averaged across Canada as a whole.
iii. Amend the Regulations to exclude free goods in “non-saleable” or “sample” package sizes, that are provided to those legally able to receive such goods pursuant to the Food and Drugs Act, from the calculation of the Average Price.
Description
This regulatory option would modify the reference to “free goods” in subsections 4(4) and 4(5) to specify that, for the purposes of the calculation of the Average Price, free goods in non-saleable package sizes provided to those legally able to receive them pursuant to theFood and Drugs Act, are not to be included in the definition of “benefit.”
Rationale
The statutory mandate of the Board pertains to patented medicines sold in Canada. If a particular package size of a patented drug is marked, for example, as “sample” and “not for sale”, and in fact is always provided free, it can be argued that this package size should not be within the Board's jurisdiction, as it is not sold in Canada.
Section 14 of the Food and Drugs Act prohibits the distribution of any drug as a sample by anyone beyond physicians, dentists, veterinary surgeons or pharmacists. The National Association of Pharmacy Regulatory Authorities (NAPRA) prohibits pharmacists from charging anything but a professional fee for their distribution.
Analysis
The extent of samples provided to the various classes of customer in non-saleable form is unknown, due to the Board's general practice of directing that they not be reported as part of the Average Price. A regulatory change formally excluding samples in non-saleable form would have little-to-no impact on the current Average Prices of patented medicines, as they would have already previously been excluded.
If the Regulations are not amended to exclude samples, then the impact of the Federal Court decision would be that all free goods, regardless of package size, must be included in the calculation of the Average Price. The resulting burden to patentees is unknown, but could be significant. As well, patentees might choose to eliminate samples rather than report them as free goods, thus potentially impeding access, on a sample/trial basis, to new medicines.
Option 4 Amend the Regulations to change “free services” to “services (free or partially subsidized)” in the calculation of the Average Price.
Description
This regulatory option would modify subsections 4(4) and 4(5) to change the words “free services” to “services (free or partially subsidized)”, thereby not artificially distinguishing between services or patient support programs simply on the basis of whether patients may pay some nominal fee for the service.
Rationale
The wording in the existing Regulations requires clarification and refinement. It is unclear why the Regulations require that “free services” be included as a benefit in the calculation of the Average Price, but do not permit services that may be subsidized by the patentee to be included, since they still represent a benefit to the patients.
Analysis
The exclusion of services which are partially or even largely subsidized by the patentee, from the calculation of the Average Price appears arbitrary. Having said this, the Board understands that services provided to patients are for the most part, if not always, free.
Option 5 Amend the Regulations to exclude “gifts” from the calculation of the Average Price.
Description
This regulatory option would delete “gifts” from the list of benefits required to be included in the Average Price calculation in subsections 4(4) and 4(5).
Rationale
The requirement to include “gifts” in the calculation of the Average Price likely stems from the past practice of the pharmaceutical industry to provide “gifts” (e.g., computers, trips, other non-medicine goods and services) to prospective and existing clients as part of a general marketing strategy. Such gifts might not have had any connection to the price of a particular patented medicine.
Analysis
Today, this is considered an unacceptable practice, and Rx&D has developed a code of conduct dictating that such gifts should not be offered. Therefore, the provision of “gifts” by a patentee should be a rare occurrence. Where they are provided, they are not likely medicine-specific and their exclusion is both reasonable and likely to have little impact on the Average Price.
Option 6 Amend the Regulations to permit the Board to disallow any or all benefits which it determines, pursuant to a public hearing, were implemented by a patentee for the purpose of reducing its liability in regard to excessive pricing in terms of the calculation of excess revenues.
Description
This regulatory option would seek to create a new regulation that gives the Board the authority, which it may use only in certain limited and specific situations, to disallow the inclusion of any benefit in the calculation of the Average Price. Specifically, the option is that the Board have this authority in the following circumstance.
i. When the Board determines that the benefits were implemented after a patentee is informed of Board's Staff's position that the price of the medicine appears to be excessive; and
ii. Pursuant to a hearing, the Board determines that the benefit was used to manipulate a price that the Board finds was excessive prior to the implementation of the benefit, in order to reduce the patentee's liability in terms of excess revenues.
Rationale
The Board's mandate is to determine whether a medicine is being or has been sold at an excessive price. If the Board determined that the patentee has used the Regulations to manipulate its price in such a way as to mitigate liability under the Act, it is proper for the Board to exercise reasonable discretion.
Analysis
In the context of its decision in the matter of LEO Pharma Inc. and the patented medicine Dovobet, the Board noted that the Average Price in Canada had been the highest in the world, and determined that the distribution of free goods (initiated after Board Staff had informed the patentee that its price appeared to be excessive) was an artificial attempt to circumvent the application of the Guidelines.
While the FCC concurred with the findings of the Board, that the distribution of free goods was not part of a genuine compassionate use program by the patentee, the judge indicated that the language of the Regulations gave him no choice but to require their inclusion in the calculation of the Average Price.
This practice of “dumping” free goods to avert liability under the Regulations could have implications or the Canadian consumer, since some markets e.g., customer classes and/or provinces/territories) ight end up paying higher and even excessive rices, while the distribution of free goods to nother market effectively reduces the overall Aerage Price for Canada to a non-excessive level.
B. Guidelines Options
Possible Changes to the CPIAdjustment Methodology for Determining the MNE Price
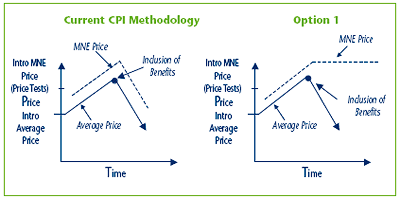
Option 1 Amend the methodology in the Guidelines for the establishment of the MNE price by using in the CPI-adjustment methodology the highest previous non-excessive Average Price, if the actual Average Price declines due to a new or increased benefit.
Description
The MNE price for a patented medicine would be calculated at introduction as it is in the current Guidelines, using the appropriate price test(s). In subsequent years, the MNE price would be calculated using the current CPI-adjustment methodology, provided that the Average Price has not decreased from the previous year.
In the event that the Average Price declines from the previous year, due to the new inclusion of, or increase in, any benefits, the calculation of the MNE price for the following year, would be based on the highest previous non-excessive Average Price, until the actual Average Price equals or surpasses the previous highest Average Price. Once this occurs, the current actual Average Price would again be used. Please see Figure 2 below for a depiction of how this option would function.
Figure 2
In cases where the new MNE price under this option could result in a significant single year price increase, some constraint would be appropriate. This could be based on a percentage maximum single year increase (e.g., not greater than 20%, or 30%, etc.) or be proportionate to the number of years the Average Price was reduced by the benefits (e.g., 100% price rebound potential if the benefit was only provided for 1 year, 33% increments if benefit persisted for 3 years, etc.).
Rationale
The Regulations specify how the Average Price must be calculated. However, the Board's statutory authority is to determine whether such an Average Price is excessive. It can be argued that if a previous Average Price was not excessive under the Board's Guidelines, then intuitively a price below this previous Average Price should also not be excessive simply as a result of the CPI-adjustment methodology.
Analysis
It is unclear how many medicines would take advantage of the flexibility inherent in this option, nor what degree of price decrease and hence level of potential price rebounds might occur. This is in part due to patentees' interpretation of the Board's policy statement, issued in the PMPRB's NEWSletter of April 2000, that allowed discretion on the part of the patentee to choose to include or exclude certain benefits from the calculation of the Average Price. Some patentees interpreted this to mean that the benefits they wished to exclude from the Average Price calculation did not even need to be reported to the PMPRB.
The application of the International Price Comparison test to ensure that the price in Canada is never the highest of the comparator countries would continue to apply. Therefore, not withstanding this option the price in Canada could never be the highest price of the seven comparator countries listed in the Regulations. If it were, the actual MNE price would then be established by the lower of the highest international price or the price generated by the proposed methodology above.
Option 2 Amend the methodology in the Guidelines for the establishment of the MNE price by using the greater of the introductory MNE price and the CPI-adjustment methodology using the highest previous non-excessive Average Price, if the actual Average Price declines due to a new or increased benefit.
Description
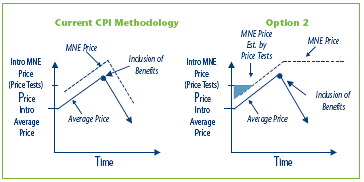
This option recognizes that benefits might be offered from the outset when the drug was first sold, and so even the Average Price in the first period of sales may include some discounts, free goods, etc. This option builds on Option 1 but adds a new element: the MNE price would be the higher of the introductory MNE price based on the introductory price test and the price resulting from the CPI-adjustment methodology.
The shaded area in Figure 3 represents the difference in potential price increase between Option 1 and Option 2.
Figure 3
Rationale
This option posits that the introductory MNE price, as established by the appropriate price test(s), would have been acceptable to the PMPRB if the medicine had actually been sold at this price at introduction. Patentees say that if they choose to offer a reduced price and therefore do not take the maximum allowable price at introduction, they should be allowed to increase their price back up to the introductory MNE price at any future time. Not to allow this would be a disincentive to the provision of any benefits in the introductory period, and could lead to drugs being introduced at the full introductory MNE price.
Analysis
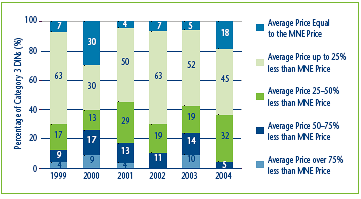
This option provides some additional pricing flexibility to those patentees that begin to sell drug products at prices below the MNE price established by the introductory price tests. Figure 4 provides some context for how Category 3 drugs8 introduced in each year from 1999-2004 were priced relative to their introductory MNE price. In 2004, 18% of drug products were priced at a level equal to the MNE price, and an additional 45% were priced within 25% of their MNE price. This indicates that in most years, the majority of Category 3 drugs introduced tend to be priced relatively close to their MNE price. However, 5% were priced between 50% and 75% below the MNE price. As with the previous option, some constraint on any single year price increase would be appropriate.
Figure 4
Distribution of Category 3 new Drug Prices relative to the Introductory MNE Price, 1999-2004
Appendix A - Overview of the PMPRB Mandate
Mandate
The PMPRB has a dual role:
Regulatory
To ensure that prices charged by patentees for patented medicines sold in Canada are not excessive thereby protecting consumers and contributing to Canadian health care.
Reporting
To report on pharmaceutical trends of all medicines, and on R&D spending by pharmaceutical patentees thereby contributing to informed decisions and policy making.
Jurisdiction
Regulatory
The PMPRB is responsible for regulating the prices that patentees charge – the factory-gate price – for prescription and non-prescription patented drugs sold in Canada to wholesalers, hospitals, pharmacies or others, in each province and territory, for human and veterinary use, to ensure that they are not excessive. The PMPRB regulates the price of each patented drug product, including each strength of each dosage form sold in Canada. This is normally the level at which Health Canada assigns a Drug Identification Number (DIN).
Health Canada assesses new medicines to ensure that they conform to the Food and Drugs Act and the Food and Drug Regulations. Formal authorization to market or distribute a medicine is granted through a Notice of Compliance (NOC). A medicine may be temporarily distributed with specified restrictions before receiving an NOC, as an Investigational New Drug or under Health Canada's Special Access Programme. Both approved and unapproved patented medicines sold in Canada fall under the PMPRB's jurisdiction.
The PMPRB has no authority to regulate the prices of non-patented drugs, and does not have jurisdiction over prices charged by wholesalers or retailers, or over pharmacists' professional fees. Also, matters such as whether medicines are reimbursed by public drug plans, distribution and prescribing are outside the purview of the PMPRB.
Under the Patented Medicines Regulations, 1994, patentees are required to file price and sales information twice a year for each strength of each dosage form of each patented medicine sold in Canada for price regulation purposes.
Patentees are also required to inform the PMPRB of their intention to sell a new patented medicine. They are not required to obtain approval of the price of a patented medicine before it is sold, but they are required to comply with the Act to ensure that prices of patented medicines sold in Canada are not excessive. In the event that the Board finds, after a public hearing, that a price is or was excessive in any market it may order the patentee to reduce the price and take measures to offset any excess revenues it may have received.
Reporting
The PMPRB reports annually to Parliament, through the Minister of Health, on its activities, on pharmaceutical trends relating to all medicines, and on the R&D spending by pharmaceutical patentees. Patentees are required to file revenues and R&D expenditures once a year for reporting purposes.
In addition to these reporting responsibilities, under Section 90 of the Act, the Minister of Health has the authority to direct the PMPRB to inquire into any other matter. Under this provision, the Minister has directed the Board to undertake two initiatives: the National Prescription Drug Utilization Information System; and monitoring and reporting on Non- Patented Prescription Drug Prices.
Appendix B - PMPRB Patented Medicine Price Review Process
In Canada, new patented medicines are assessed by Health Canada to ensure that they conform to theFood and Drugs Act and the Food and Drug Regulations with respect to safety, quality and efficacy. For a new medicine to be marketed or distributed in Canada it must be granted a Notice of Compliance (NOC). A medicine may also be sold with specified restrictions before receiving an NOC, either as an Investigational New Drug (IND) or under Health Canada's Special Access Programme (SAP). SAP drugs have not been approved for sale in Canada but a physician may request authority to obtain the drug from outside Canada for a specific patient.
For the jurisdiction of the PMPRB to crystallize, a medicine must be both patented and sold in Canada. However, once a patent for a medicine has been issued, it is the policy of the Board to retroactively review the price at which the medicine was first sold during the patent pending period.
While patentees are required to inform the PMPRB of an “intention to sell” a new patented medicine, the PMPRB's price review process is ordinarily triggered by the patentee filing information on the identity of the medicine once the medicine is actually patented and being sold in Canada. At specific times and for specific periods as laid out in the Patented Medicines Regulations, 1994 (Regulations), the patentee is also required to report price and sales data for each drug product, reported at the level of the Drug Identification Number (DIN9).
The scientific review requires the product monograph of the drug product under review. In addition, a patentee may make a submission regarding how the medicine should be categorized (line extension of an existing drug; breakthrough or substantial improvement; or moderate, little or no improvement), along with proposed comparators, dosage regimens and supporting clinical studies.
The scientific review involves an evidence-based process that determines the appropriate category for the drug, appropriate therapeutic comparators and comparable dosage regimens. It does not consider any pricing information relating to the new product. All new active substances are referred to the Human Drug Advisory Panel (HDAP) to review and evaluate clinical trial and other scientific evidence. Based on the categorization of the drug, Board Staff conducts the appropriate price tests as prescribed by the Guidelines.
The price tests generally use a single Average Price for all of Canada (although the Board retains the right to review Average Prices in any market). The Average Price is calculated for each drug product (identified at the level of the DIN) by dividing the total net revenue (i.e., the sum of revenues from each class of customer in each province/territory) by the total units sold (i.e., to all classes of customer in each province/territory).
Board Staff compares the Average Price for each DIN to the Maximum Non-Excessive (MNE) price established using the price tests laid out in the Guidelines, which were developed pursuant to the price factors in subsection 85(1) of the Patent Act (Act), to determine if the price of the drug is excessive.
If the patentee's Average Price is not above the MNE price established by the appropriate price tests, the patentee's price is deemed to be “within the Guidelines.” The Average Price in the introductory period then sets the benchmark for future monitoring of prices. In future years, the MNE price is determined by the lesser of the MNE price established through the CPI-adjustment methodology or the highest price at which the medicine is sold in the seven comparator countries listed in the Regulations.
If the price at which the medicine is being sold is considered excessive and triggers the investigation criteria set out in the Guidelines, Board Staff commences an investigation. There are three ways to resolve an investigation:
- further submissions by the patentee and information obtained by Board Staff result in a determination that the price is within the Guidelines;
- the patentee voluntarily agrees to reduce the price and pay back any excess revenues; or
- the matter is referred to the Board Chairperson who decides whether it is in the public interest to hold a public hearing.
Appendix C - Bibliography
Legislation, Regulations and Guidelines Documents
Patent Act
Patented Medicines Regulations, 1994
PMPRB's Compendium of Guidelines, Policies and Procedures
Patentee's Guide to Reporting
PMPRB NEWSletters
Documents from the Review of the Excessive Price Guidelines*
* For ease of reference, information has been set-up in a chronological order.
Discussion Guide for the Consultations on the Board's Excessive Price Guidelines; Released: May 2006.
Stakeholder Comments Received on the May 2006 Discussion Guide
Participant Kit for the Face-to-Face Consultation Meetings in November 2006
Summary Reports from Face-to-Face Consultation Meetings in November 2006
PMPRB Stakeholder Communiqué, May 31, 2007
Board's Bilateral Meetings with Stakeholder Groups in September 2007 – List of Participants by Meeting
Board's Bilateral Meetings with Stakeholder Groups in September 2007 – Written Comments Received from Participants
1 The Health Portfolio is comprised of Health Canada and five agencies: the Assisted Human Reproduction Agency of Canada, the Canadian Institutes for Health Research, the Hazardous Materials Information Review Commission, the Patented Medicine Prices Review Board and the Public Health Agency of Canada.
2 All background documentation from previous consultations on the Guidelines is posted on the Board's Web site:
3 For more details on the implications of the FCC decision, see the April 2007 NEWSletter article posted on the Board's Web site:
4 Note: Work on these proposed regulatory amendments began in 2005 (prior to the Guidelines review) and was intended to improve the efficiency and effectiveness of the current price review process. For stakeholder comments on the second pre-publication of proposed regulatory changes published in Canada Gazette, Part I, see the Board's Web site:
5 A DIN (or Drug Identification Number) is assigned by Health Canada at the time a Notice of Compliance (NOC) is issued, which permits the drug to be sold in Canada. This DIN is applied at the level of each unique strength and dosage form of a medicine. PMPRB generally reviews the prices of drugs at the DIN level. Even in cases where an NOC has not been issued, but the medicine is sold (for example under Health Canada's Special Access Programme), the PMPRB will review the price of each unique strength and dosage form.
6 Under the Patented Medicines Regulations, 1994, the comparator countries are France, Germany, Italy, Sweden, Switzerland, the United Kingdom and the United States.
7 An NOC/c is authorization to market a drug (i.e. a Notice of Compliance (NOC)), with the policy condition that the sponsor undertake additional studies on the clinical benefit of the product.
8 A Category 3 drug is a new DIN of a non comparable dosage form of an existing medicine, or the first DIN of a new chemical entity that provides moderate, little or no therapeutic advantage over comparable existing DINs. Typically, the MNE price is determined by the highest price of other drugs sold in Canada that are considered therapeutically comparable.
9 A DIN (or Drug Identification Number) is assigned by Health Canada at the time a Notice of Compliance (NOC) is issued, which permits the drug to be sold in Canada. This DIN is applied at the level of each unique strength and dosage form of a medicine. PMPRB generally reviews the prices of drugs at the DIN level. Even in cases where an NOC has not been issued, but the medicine is sold (for example under Health Canada's Special Access Programme), the PMPRB will review the price of each unique strength and dosage form.